I am a Computational Associate at the Broad Institute of MIT and Harvard, Massachusetts General Hospital, who is focused on developing and applying interpretable computational methods to biomedicine domain. My goal is to bridge the gap between clinicians and computational researchers building interpretable and scalable algorithms that handle multimodal data such as Electronic Health Records, -omics and flow cytometry data enhancing field of personalized medicine.
As aspiring PhD student who is looking to study Biomedical and Computational Biology, I want to form active clinical collaborations to help develop accurate models for handling multi modal biological data. These models will be integrated into hospital workflows to aid clinical decision making and to tailor patients’ care to their unique clinical and genomic traits.
- Bridging wet lab discoveries to clinical applications
- Biomarker discovery and validation platforms
- Computational algorithms
BSc in Biophysics, 2022
Lomonosov Moscow State University
Experience
Projects:
- RNA-Seq analysis and Tandem Mass Tag Proteomincs analysis:
- Developing and maintenaning computational pipelines using publicly available and in-house developed tools for the analysis of experiment data.
- Monitoring statistical aspects of study/experiment design, identification of appropriate tests and models, data analysis and visualization, assuring quality of data.
- Worked with wet lab biologists helping in data analysis and visualizations.
Projects:
- Machine learning models deconvolution:
- Assessed performance of machine learning models for tumor micro-environment deconvolution using statistical methods.
Projects:
- Automated flow cytometry pipeline:
- Led a project to implement an innovative automated flow cytometry data analysis pipeline using gradient boosting models, significantly reducing (1400-fold speedup) data analysis time for wet bench researchers.
- By automating the data processing and classification steps, the pipeline improved workflow efficiency and allowed for quicker access to critical insights.
- This work was used for multiple projects within the laboratory.
- Differential gene expression methods benchmarking:
- Worked with computational biology group from the university of Warsaw on a large collaborative project focusing on the analysis of single cell RNA-seq data from hundreds of patients.
- Benchmarked traditional approaches for differential gene expression methods vs deep learning-based approaches.
- The results of this work were essential for downstream tasks in this project.
- The project is still in revision-stage (2025)
- Single cell spatial transcriptomics analysis:
- Researched spatial transcriptomics platforms (Nanostring CosMx, 10x Xenium) that helped bringing these technologies to the laboratory.
- Utilized various best-practice computational tools and techniques to decipher spatial patterns and identify potential biomarkers associated with pulmonary diseases, assessed publicly available datasets.
- Single cell RNA-seq data analysis:
- Analyzed single-cell data from pulmonary samples to gain insights into cellular heterogeneity and molecular changes in disease conditions.
- Employed bioinformatics tools to preprocess and interpret single-cell RNA sequencing data, revealing critical cellular subpopulations and their functional roles.
Projects:
- Cell type deconvolution of cancer- associated fibroblasts in bulk RNA-seq data using machine learning methods with the goal to improve outcome prediction in cancer patients:
- Led development of a project classifying fibroblasts in tumor microenvironments using machine learning and gene expression signatures, which resulted in my thesis.
- Presented work at OpenBio 2022.
- Work on this project was continued and results were submitted to AACR 2024.
- Machine learning models validation:
Developed a script to validate machine learning models that deconvolve sample cells’ percentages based on sequencing data. - Enhanced cross-team communication:
Developed standard report form of RNA samples analysis to share results efficiently across multiple bioinformatics teams. - Reproducible programming and RNA-seq analysis:
Performed code debugging, analysis, and filtration of RNA-seq lab samples.
Relevant courses and workshops
Took multiple workshops on various topics related to the use of machine learning in bioinformatics, led by academia leaders:
- Machine Learning for Molecule Data (Jose Miguel, University of Cambridge),
- Machine Learning in Protein Structural Bioinformatics (Cryo-EM and protein docking) (Daisuke Kihara, Purdue University),
- Biomedical text mining and its application (Shannkai Yan, National Institutes of Health),
- DNABERT: BERT models for genome DNA language (Ramana Davuluri, Stony Brook University).
Went through extensive selection (100/5000 people) and took classes from academia and industry leaders in:
- Machine learning (A. Djakonov, Lomonosov Moscow State University),
- Data engineering (P. Klemenkov, NVIDIA),
- Statistics (V. Panov, Computer science department, Higher School of Economics),
- Linear algebra (I. Oseledets, Skoltech),
- Linux, Bash, and Git (A. Trunov).
Projects

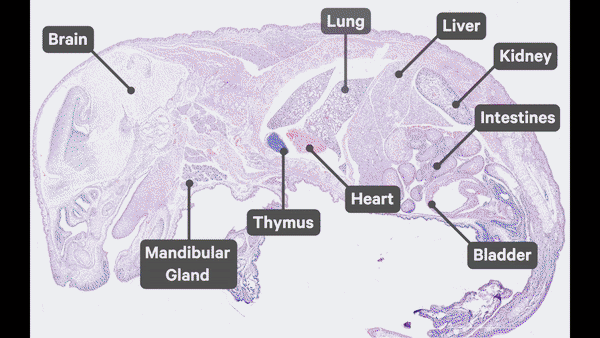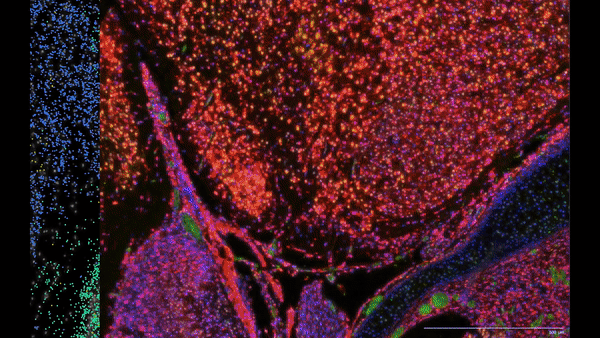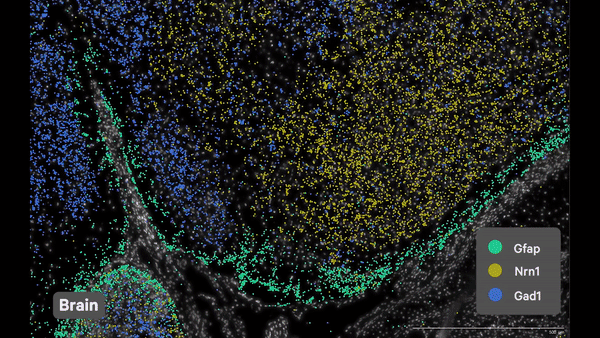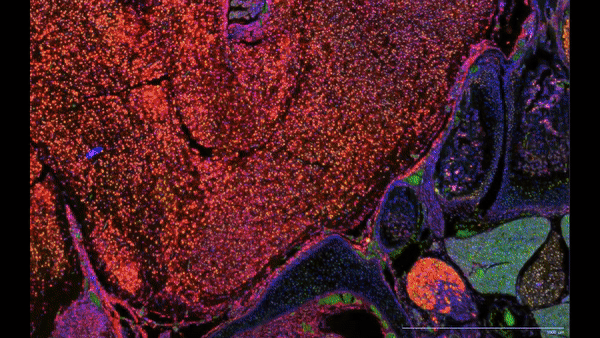
Manuscripts In Progress
Hercher T.W., To T.L., McCoy J., Wu A., Bratchikov S., Durham T., Vantaku V.R., Parangi S., Mootha V.K. (in preparation) Robust Generation of Oxygen at the Surface of Human Cells via Plasma Membrane-Targeted SNORCL
Fenske S.W., Peltekian A., Kang M., Markov N.S., Zhu M., Grudzinski K., Bak M.J., Pawlowski A., Gupta V., Mao Y., Bratchikov S., Stoeger T., Rasmussen L.V., Choudhary A.N., Misharin A.V., Singer B.D., Budinger G.R.S., Wunderink R.G., Agrawal A., Gao C.A., and the NU SCRIPT Study Group (medRxiv 2024.06.28.24309547 Scientific Reports, Accepted after revision) Developing and validating a machine learning model to predict successful next-day extubation in the ICU
Posters & Talks
Posters
Machine Learning Classifier for Automated and Scalable Analysis of Clinical Flow Cytometry Samples Bratchikov S. et al
- Presented at: 2023 Systems Biology for Infectious Diseases Annual Conference, Northwestern University
- Presented at: 2023 Molecular Mechanisms of Lung Disease Annual Meeting, University of Helmholtz - Northwestern University Conference
- Presented at: 2023 The 17th Annual Lewis Landsberg Research Day, Northwestern University
Talks
RNA Expression-Level Heterogeneity in Cancer-Associated Stromal Cell Populations within the Tumor Microenvironment.
- Presented at OpenBio 2022 conference
Contact
Feel free to contact via email:
- sbratchi@broadinstitute.org
- 415 Main St., Cambridge, MA 02142